-
 Get UpdatesGet notified on education trends, resources, and growth opportunities!
Get UpdatesGet notified on education trends, resources, and growth opportunities! -

-


 Click it and Unblock the Notifications
Click it and Unblock the Notifications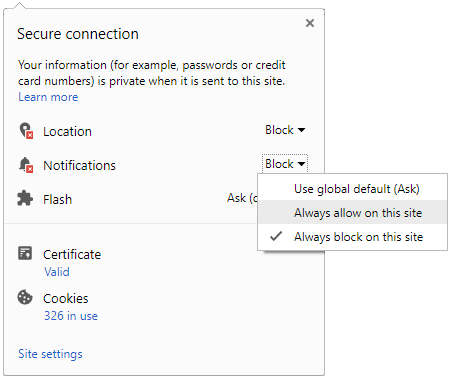
IIT Madras Develops DeepPPAPredMut: AI Tool For Protein Mutation Prediction
IIT Madras has developed DeepPPAPredMut, an AI-based tool that predicts the impact of protein mutations. This user-friendly innovation streamlines research, offering a faster and cost-effective alternative to existing methods in biotechnology.
Scientists at the Indian Institute of Technology (IIT) Madras have made a significant advancement in the field of biotechnology with the development of 'DeepPPAPredMut', an artificial intelligence-based tool designed to predict the impact of mutations on proteins. This innovation, highlighted in the esteemed "Bioinformatics" journal, stands out by offering a faster and more cost-effective alternative to traditional methods, which are often hindered by their laborious, time-consuming, and expensive nature. The tool, equipped with a user-friendly web interface, aims to make this complex analysis accessible to researchers worldwide, marking a pivotal step forward in protein study.

Understanding the behavior of proteins, which are fundamental to numerous biological functions, is crucial in the realm of science. Proteins are involved in a variety of cellular processes, including cell signaling, immune responses, and the cell cycle, serving roles as enzymes, structural components, and more. However, the stability and functionality of protein-protein complexes can be compromised by mutations, potentially leading to a range of diseases. This brings to light the importance of studying protein mutations and their impacts on biological processes.
"Hence, there is a dire need for computational approaches to predict the changes in binding-free energy caused by mutations within protein-protein complexes," explained M Michael Gromiha, a Professor at the Department of Biotechnology, IIT Madras. He further elaborated on the limitations of current methods, which are categorized into 'structure-based' and 'sequence-based' approaches. Structure-based methods are often hampered by the scarcity of experimentally known structures for protein-protein complexes, while sequence-based methods, despite their development for predicting changes in binding affinity due to mutations, have their own set of challenges.
Delving into the realm of protein interactions, Rahul Nikam from the Department of Biotechnology at IIT Madras shed light on how these interactions are foundational to many cellular functions. Mutations within protein-protein interactions can disturb these functions, leading to various diseases. "With the evolution of protein structure prediction methods like AlphaFold2 and the availability of extensive experimental affinity data, there is a pressing need for updated computational tools that can efficiently predict changes in binding affinity caused by mutations in protein-protein complexes," Nikam remarked. This statement underscores the need for innovations like 'DeepPPAPredMut', which not only addresses existing gaps but also capitalizes on the latest advancements in protein structure prediction and data availability.
'DeepPPAPredMut' takes a step beyond existing methodologies by offering a sequence-based approach that requires only the protein sequence and mutation details from the user to predict changes in binding affinity within the protein complex. This AI-driven tool represents a significant leap in our ability to understand and mitigate the effects of protein mutations, providing a more streamlined and accessible means for researchers to explore the complex dynamics of protein interactions and their implications for health and disease.
In conclusion, the development of 'DeepPPAPredMut' by researchers at IIT Madras signifies a major advancement in the study of proteins and their mutations. By combining the power of artificial intelligence with a user-friendly interface, this tool not only surpasses traditional methods in efficiency and cost-effectiveness but also opens new avenues for research in biotechnology and medicine.
More From Career India











- Block for 8 hours
- Block for 12 hours
- Block for 24 hours
- Don't block
- Male
- Female
- Others
- Under 18
- 18 to 25
- 26 to 35
- 36 to 45
- 45 to 55
- 55+